 |
||
|
|
runMultiTCW Guide | |
|
||
Note, familiarity with singleTCW is essential, as MultiTCW projects are created by merging existing sTCW projects.
Contents
- Overview
- Running the demo - Highly recommended to do this.
- The four steps
- Additional Details
- References
Overview | Go to top |
| mTCWdb | multiTCW database |
| sTCWdb | singleTCW database |
| NT | Nucleotide (transcript, gene) |
| AA | Amino acid (translated ORF, protein) |
| NT-sTCWdb | singleTCW created from NT sequences. |
| AA-sTCWdb | singleTCW created from AA sequences. |
- Input:
- Two or more sTCW databases. The sequences, annoDB hits, GOs, TPM and DE p-values are imported to the mTCWdb.
- For NT-sTCW, both the nucleotide sequences and their translated ORFs will be imported into the mTCWdb.
- The results are the best if:
- The sTCW databases are annotated the same.
- The conditions names are exactly the same (when applicable). For example, if two species both have counts for the tissue type 'leaf', the condition name provided in runSingleTCW must be the same for both (e.g. leaf), though the name is case-insensitive.
- The DE column names are exactly the same (when applicable), as in the previous point.
- Computation:
- Compare the AA sequences using
diamond orblast , and the NT sequences withblastn . - Compute one or more sets of clusters using the following methods: BBH (bi-directional best hit), Closure, Best hit, orthoMCL1, and/or user-supplied clusters.
- For a NT-mTCW database created from only NT-sTCW databases, statistics such as Ka/Ks2, synonymous, etc are computed.
- The clusters are scored.
- Compare the AA sequences using
Software Requirements and Installation | Go to top |
Running the Demo
This section is essential for learning how to useCreate the three 'ex' sTCWdbs: Using
|
|
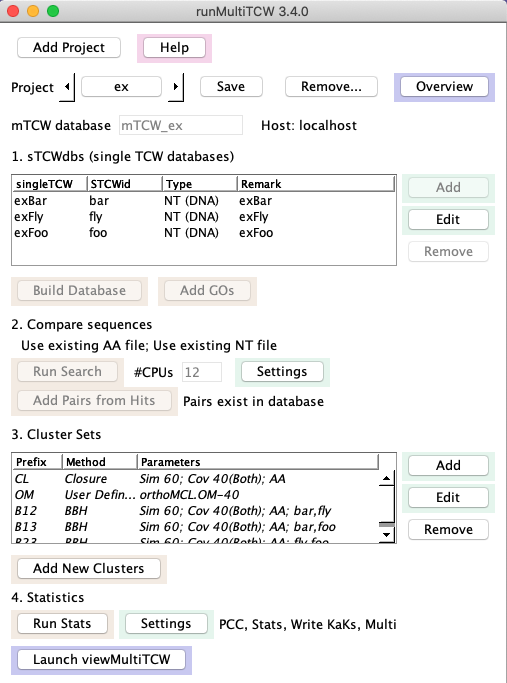
Create mTCW_ex: A skeleton
→Overview: The overview will look similar to this Overview (counts may vary slightly). In this example, the |
|
To try mixing NT-sTCWdb and AA-sTCWdb as input, create a mTCWdb with demoTra, demoAsm and demoPro. You can
also experiment with only AA sTCWdbs by first making protein sTCWdbs from the ORF files of
exBar, exFoo and exFly (use the project's
The four steps | Go to top |
Top three rows
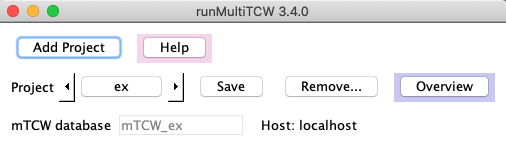
|
| A popup window will appear where you enter the project name. On 'OK', the
following occurs:
(1) A project directory will be created under (2) A file called (3) The database will be the same name with the prefix 'mTCW_' added. |
| A pop-up window that provides similar information to this UserGuide. | |
| The drop-down lists all sub-directories under | |
| The entered information is saved in | |
| Once you have selected a project, you can select | |
| By default, the mySQL name will be |
|
|
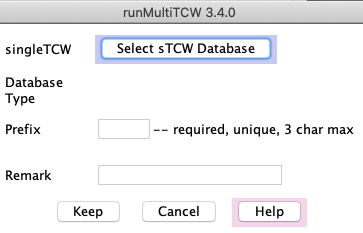
1. sTCWdb (single TCW databases)
A multiTCW database can be created with a mix of NT-sTCWdbs and AA-sTCWdbs.|
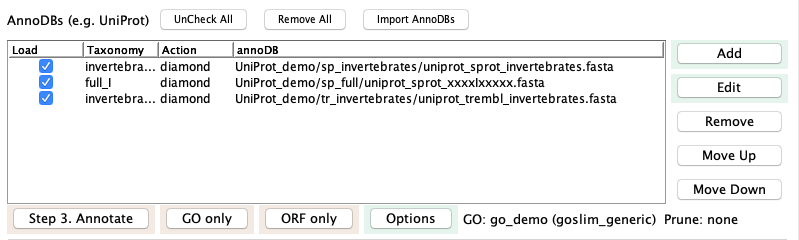
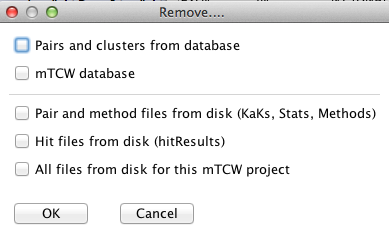
Click the Click The 'prefix' is only used in the Method files, so it does not matter what it is as long as its unique. The remark can be anything, and can be added/changed after the database is created. Avoid special characters such as quotes. When you select Repeat to add all the sTCWdbs you want to compare, then execute |
|
2. Compare sequences
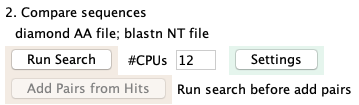
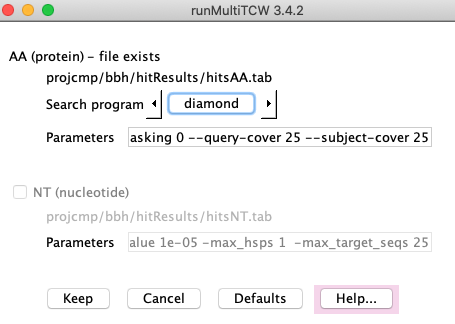
The image on the right is the On the Once the search is run, you can no longer change the parameters; if you want to change the
parameter and re-run the search, remove the hit files using the main panel |
|
Add Pairs from Hits
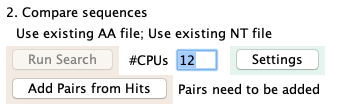
| On the main window, loads the pairs from the hits result file. This step also assigns the shared description, which is the same algorithm as used for the Majority Hit for clusters. The only difference is that if there is no shared hit, the hitID will be "*NoShare" whereas clusters always are assigned an annotation. |
3. Cluster Sets
Methods
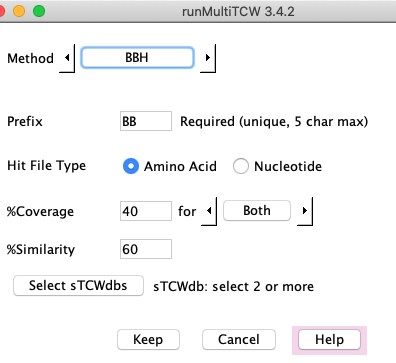
ClickBBH
The BBH finds the bi-directional best hit based on hit e-value.
It uses the hits that were loaded
into the database with
|
|
%Coverage:
- This is computed by:
Cov = (alignment_length/sequence_length_1)*100; this can give a %Coverage>100 if there are many gaps and the alignment is the same length as the sequence. - You can choose "Either", which requires that either
Cov1 >%Coverage ORCov2 >%Coverage. If you choose "Both", thenCov1 >%Coverage ANDCov2 >%Coverage. For example, for an alignment:Seq1 ------------------------------- Seq2 ----------
This will pass the filter %Coverage>=80 if "Either" is selected, but not "Both".
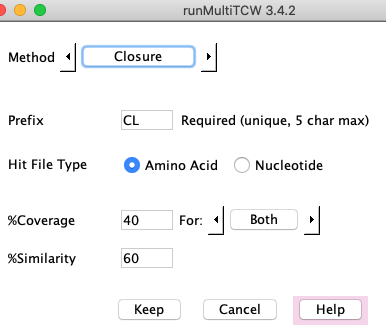
Closure
Closure has the same %Coverage and %Similarity parameters as the BBH algorithm, and also uses the hits from the database.
It creates clusters as follows:
|
|
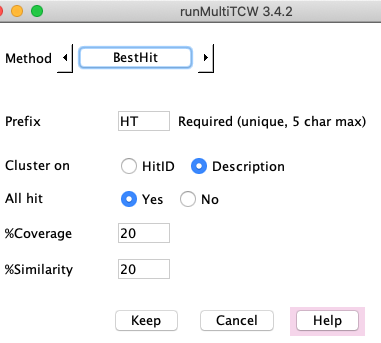
BestHit
Clusters are formed using the best annotation for each sequences, as follows
(protein hit or pair hit indicates a DIAMOND/BLAST result):
|
|
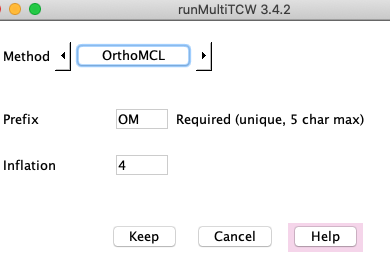
OrthMCL
|
|
|
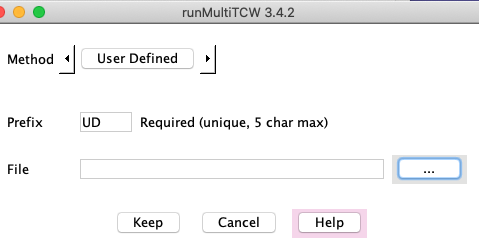
User-defined clusters
Create a file specifying the groupings, and the interface simply uploads that file. Hit results
are not used. The group file has the following format:
.. D26: tra|tra_030 tra|tra_184 tra|tra_094 pro|pro_100 D27: tra|tra_045 tra|tra_209 pro|pro_011 ...Each line starts with "DN", where N is the group number, and then has a space-separated list of the sequences in the group, prefixed by the project prefix that you entered when you set up the mTCW. The is the same file format used by all methods. |
|
Majority hit per cluster
For all the cluster methods, a majority hit is assigned to the cluster using the same rules are for the Pairs Annotation. The only difference is that the cluster will always be assigned an annotation even if there is only one sequence with the given annotation. The rules in the context of clusters:- For a cluster, a list of 'Best Anno' is created where they are
modified as follows:
- The words "probable", "punitive", or "predicted:" are removed.
- The first two words are extracted.
- The annotation is saved in lower case.
- The number of times an abbreviated annotation is found for a sequence in the cluster is counted (counted only once per sequence no matter how many times it is found in the sequence list).
- The annotation that has the highest count is assigned to the cluster. If there is a tie, then is further sorts on E-value, whether is it SwissProt and number of GOs. The cluster is also assigned an associated hitID that has the best E-value.
- A sequence can have a 'Best Anno' of "uncharacterized" if it has no good annotation hits. This will not be used unless all sequences in the cluster only have "uncharacterized".
- If none of the sequences annotation, the hitID will be "*Novel".
| For example, in the cluster on the right, all three sequences have the annotation "chromatin-remodeling complex" in their annotation list. However, the second sequence does not have the annotation "ATP-dependent chromotin" in its annotation list. | 
|
The %Hit is computed, which is the percentage of sequences with the hit. This hit is not necessary the "Best" hit for any of the sequences.
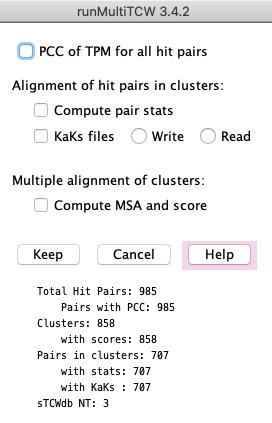
4. Run Stats
The statistics are broken into four sections:
|
|
Details on running KaKs_calculator
After the KaKs files have been created using - Exit
runMultiTCW or use a different terminal window. - Change directories to
projcmp/<project-name>/KaKs . There will be multiple files with the nameoTCWn.awt where n starts at '1'. They each have pairs of aligned sequences minus the gaps. There is also a file calledrunKaKs which has the commands to run theKaKs_calculator on each file, e.g./Users/cari/Workspace/TCW_4/Ext/mac/KaKs_Calculator -i oTCW1.awt -o iTCW1.tsv -m YN & /Users/cari/Workspace/TCW_4/Ext/mac/KaKs_Calculator -i oTCW2.awt -o iTCW2.tsv -m YN &
The only reason the method "YN" is used here is because it is fastest; this should probably be changed. The default method is "MA", which will be used if you remove the "-m YN" on all lines. Alternatively, you can edit the file to replace the "YN" with a different method. To view the different methods, run (replace 'mac' with 'linux' if you are on a linux machine):./Ext/mac/KaKs_Calculator -h
- From the command line, type:
sh runKaKs . - Read the KaKs files into the mTCWdb:
- If you have exited
runMultiTCW and start it back up, the label beside the pair statisticsSetting will say "Read KaKs". Just selectRun Stats and the files will be read. - If you had not exited
runMultiTCW but ranrunKaKs from another terminal window, the label will still say "No action to be performed". SelectSettings , selectRead KaKs files andKeep . NowRun Stats .
- If you have exited
Details on MSA score and using MstatX
By default, Score1 is the Sum-of-Pairs. The Sum-of-pairs score compares each two characters in the column,
where there are 22 possible characters (20 amino acids, gap '-', and leading/trailing space ' ').
The comparison scores are: (aa,aa) is the BLOSUM68 score, (aa,'-') is -4, (aa,' ') is -1, ('-','-') is 0, (' ',' ') is 0.
The cluster value is the sum-of-pairs/#comparisons, where a higer score is a better score. (see note below).
Score2 is "Wentropy", which is copied
directly from the
The
./runMultiTCW -M1 <method> -M2 <method>where M1 changes Score1 and M2 changes Score2. For example:
./runMultiTCW -M1 tridentwill cause Score1 to be Trident and Score2 will be the default Wentropy computation.
Update scores: The scores can be changed after ALL the MSAs are computed, as follows:
Check
For developers: you can add your own method to the
NOTE: The Wentropy or one of the other mStatX statistics should be valid to use in a
publication. Do not use the Sum-of-Pairs score in publication unless cleared with a statistician. The Sum-of-pairs
score is useful on the MSA display in
Additional details | Go to top |
Timings
The following times are from the log files for building an mTCW database with three NT-sTCWdbs.| Step | Time | Added |
|---|---|---|
| Build Database | 5h:0m:36s | 138,907 sequences |
| Add Pairs | 2h:3m:04s | 454,568 pairs |
| Add New Clusters | 1h:23m:05s | 46,831 clusters |
| Run Stats | 1h:33m:15s | 116,109 alignments |
The longest task is to
The search program (e.g. blast) is run on
Trouble shooting | Go to top |
A file called
View/Query with
| Go to top |
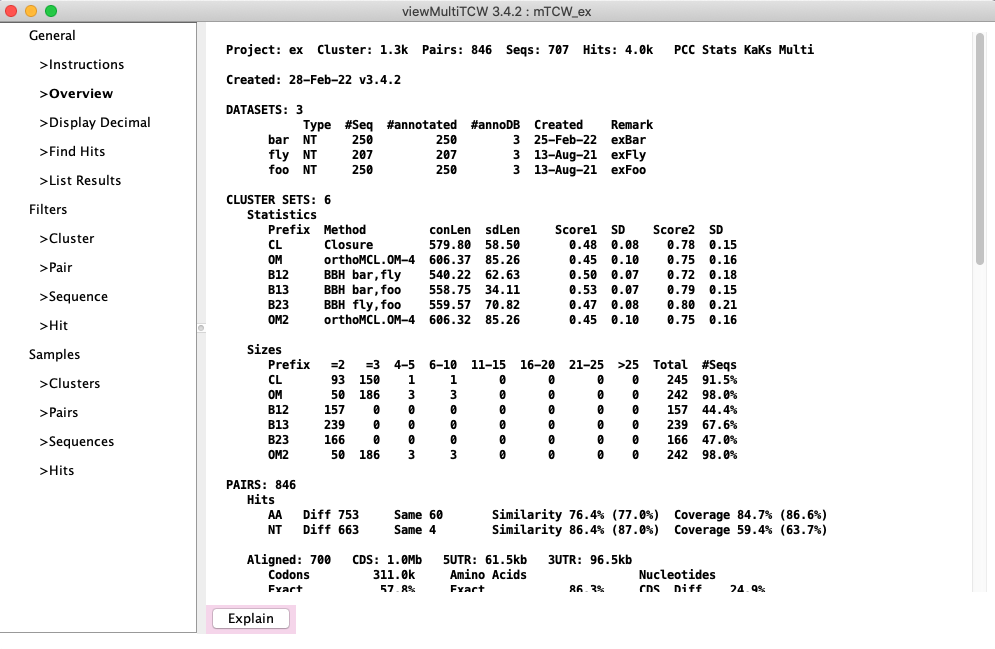
The clusters can be viewed by either:
|
|
References | Go to top |
- Li, L., Stoeckert, C.J., Jr. and Roos, D.S. (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res, 13, 2178-2189.
- Zhang Z, Li J, Xiao-Qian Z, Wang J, Wong, G, Yu J (2006) KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Geno. Prot. Bioinfo. Vol 4 No 4. 259-263.
- Katoh K, Standley DM (2013) MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution Vol 30, Issue 4 772:780
- Guillaume Collet (2012) https://github.com/gcollet/MstatX.
- Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792-1797.
| Email Comments To: tcw@agcol.arizona.edu |