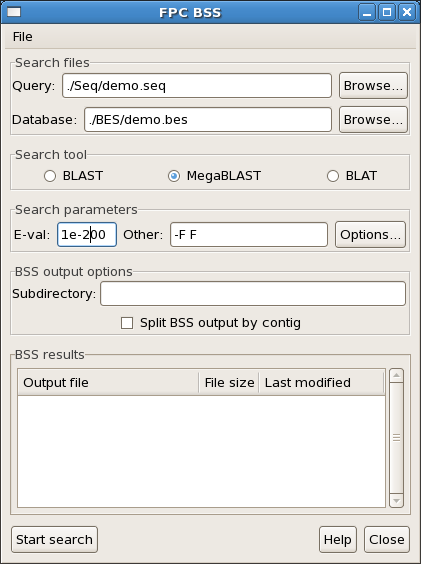
allows easy alignment and visualization of a draft sequence assembly against
an FPC clone map of the same species. This alignment permits you to check both sequence and FPC
assemblies and locate possible sequence or contig merges.
This demo illustrates all four possibilites.
Note that BES must be named as clonename+r or clonename+f. In other words,
if your clone name is a0001B13, then the BES would be a0001B13r and a0001B13f. (A ".r" and
".f" ending will also work.)
Note that for the flat representation of the draft sequence, the software does not specifically
recognize the gaps notated by N's. If your sequences
have gaps which you wish to see visually in the display, you must use the scaffolded
representation.
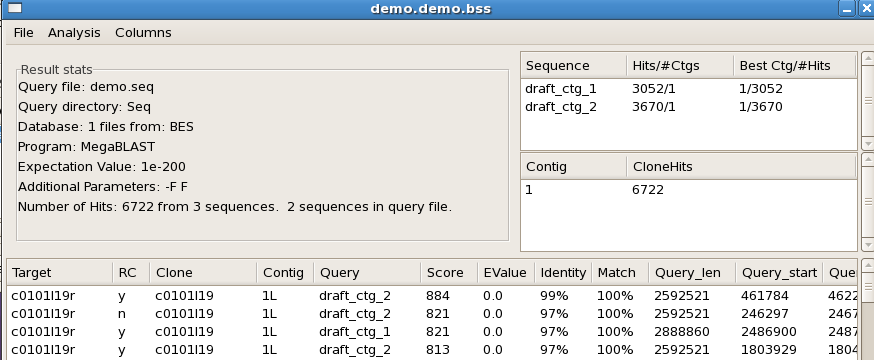
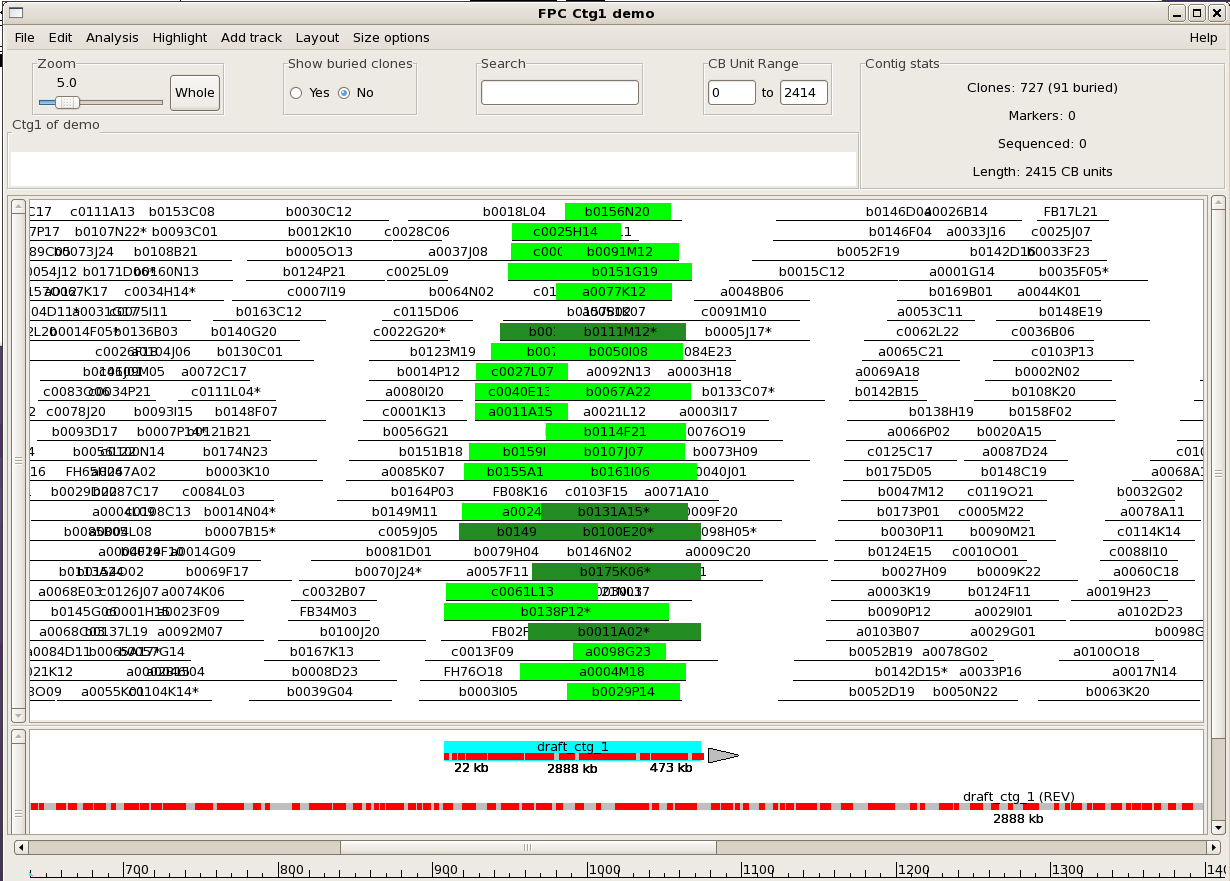
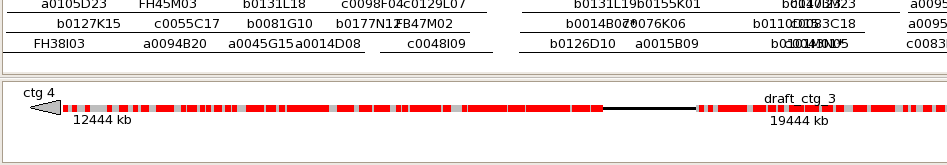
Once the alignments are computed, they are displayed in a new "sequence track" within
the contig view (for a demo on manipulating the FPC tracks, see Track Tutorial").
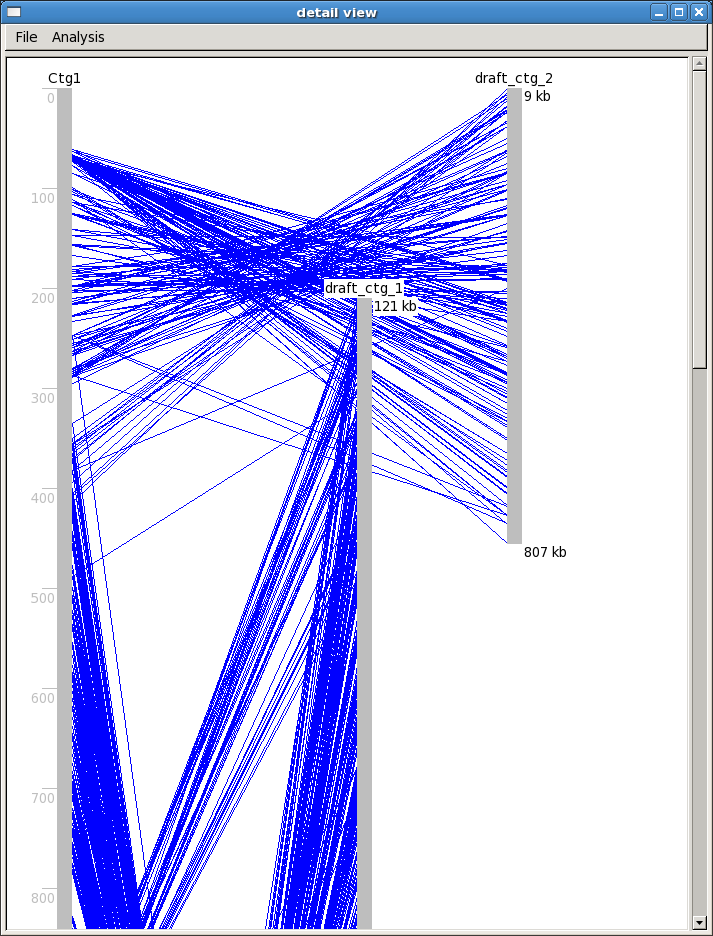
A "detailed alignment" view is also available, which opens a separate window showing
each individual BES hit between the sequence and the FPC contig.
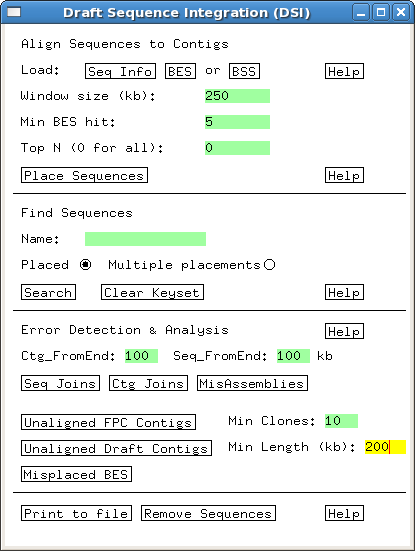
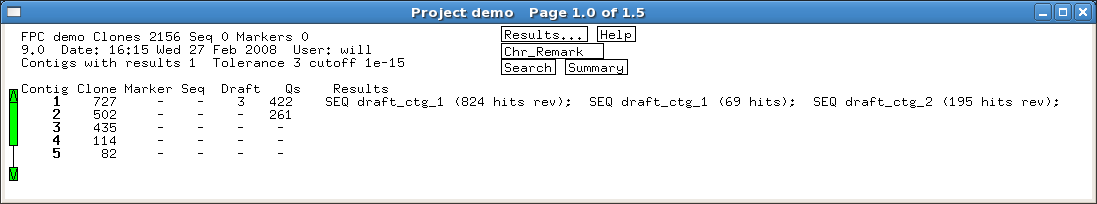
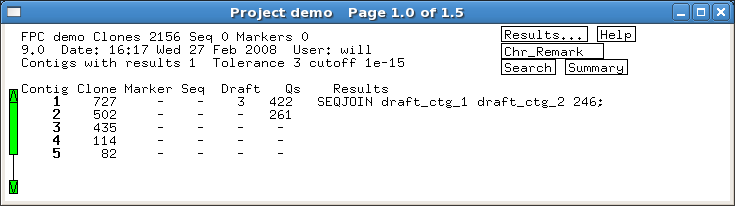
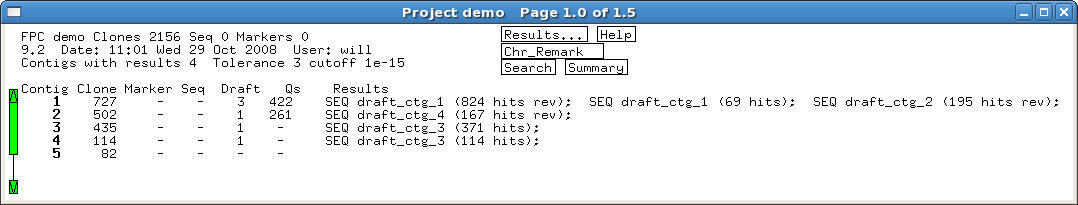
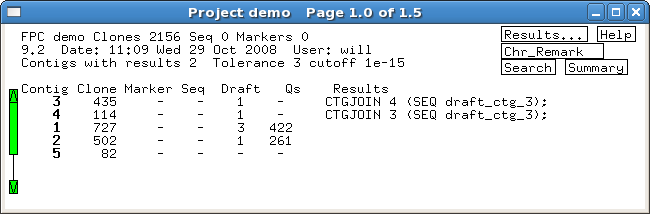
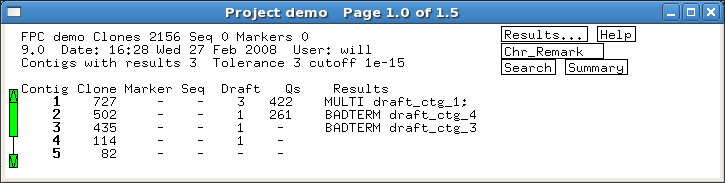
Several built-in error detection functions
are provided to extract information from the alignments, including potential
misassemblies, sequence joins, contig joins, sequences and contigs which did not align,
and BESs whose location conflicts with an alignment.
To begin on the demo, download the package seqdemo.tar.gz.
Save it to an empty directory and unpack it with the commands
gunzip seqdemo.tar.gz
tar xvf seqdemo.tar
A new directory "seqdemo" is created. Change to this directory and verify that
you see the following listing of files: