
|
| AW - Allele Workbench | ||
| |||
| Download |
|
AGCoL Pipeline.tar.gz:
Pipeline scripts, external programs, pipeline demo files
The pipeline demo files are only available from the AGCoL website due to their size (407M). However, the pipeline perl scripts are available from the AW Github source. | |
| Github AW_1_1.tar.gz: runAW, viewAW, and the interface demo files |
|
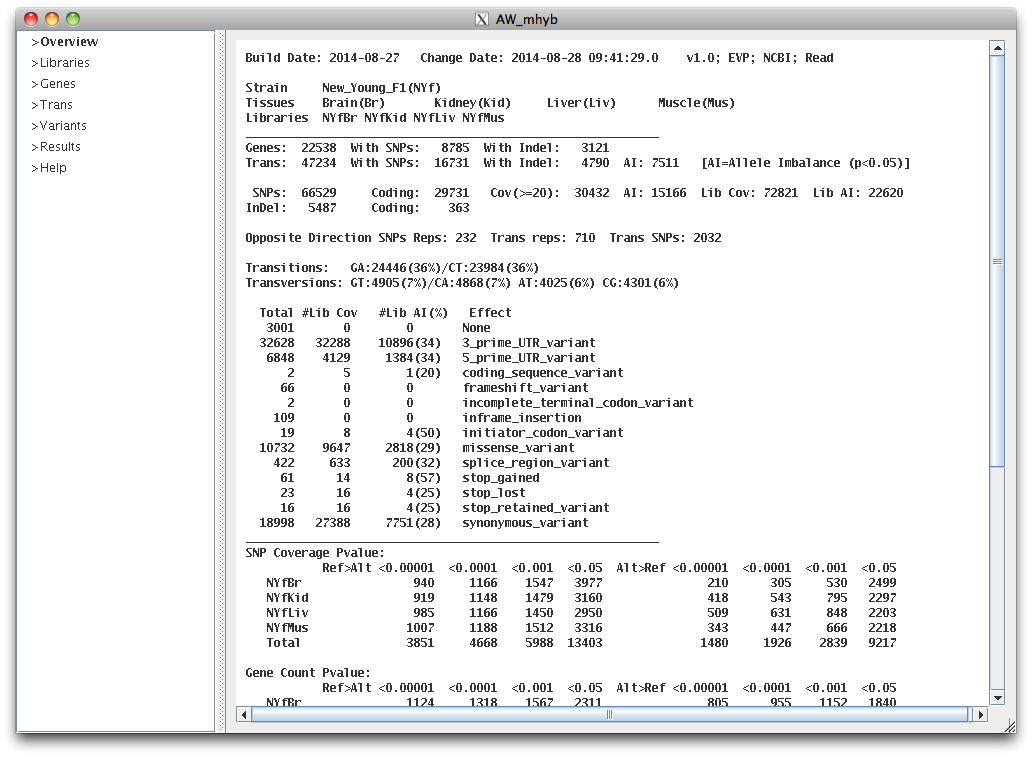
viewAW overview
|
Funding
This project is funded in part by National Science Foundation grants DBI-1265383 and IOS-1248090.
NSF Disclaimer: Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.